| Table of Contents |  |
|
Case Report
| ||||||
| Coexistence of Mayer–Rokitansky–Küster–Hauser syndrome with Turner syndrome: A case report | ||||||
| Nik Rafiza Afendi1, Hoo PS2, Mas Irfan Jaya Mahamooth3, Ahmad Amir Ismail1, Rahimah Abdul Rahim1, Ahmad Akram Omar1, Adibah Ibrahim1 | ||||||
|
1Senior lecturer, Obstetrics & Gynecology department, School of Medical Science, University Sains Malaysia, Kota Bharu, Kelantan, Malaysia.
2Clinical lecturer, Obstetrics & Gynecology department, School of Medical Science, University Sains Malaysia, Kota Bharu, Kelantan, Malaysia. 3Trainee lecturer, Obstetrics & Gynecology department, School of Medical Science, University Sains Malaysia, Kota Bharu, Kelantan, Malaysia. | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] |


| How to cite this article: |
| Afendi NR, Hoo PS, Mahamooth MIJ, Ismail AA, Rahim RA, Omar AA, Ibrahim A. Coexistence of Mayer-Rokitansky-Küster-Hauser syndrome with Turner syndrome: A case report. Edorium J Gynecol Obstet 2017;3:5–8. |
|
Abstract
|
|
Introduction:
Turner syndrome (gonadal dysgenesis) is an important cause of short stature and primary amenorrhea in young women. It is the most common sex chromosome abnormality in females and occurs approximately 1/2500 live births, caused by loss of part or all of an X chromosome. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome on the other hand affects 1/4500 births and rarely associated with gonadal dysgenesis.
Case Report: We report a case of a 19-year-old female presented with primary amenorrhea and short stature. On examination, patient’s height was 139 cm, and she had no pubic or axillary hair. Breast examination revealed breast buds only. She subsequently underwent MRI scan which showed absent vagina, uterus and cervix, while karyotyping was consistent with Turner syndrome. Conclusion: An association between these two conditions is very rare and appears to be coincidental, independent of chromosomal anomalies although a few theories have been suggested. Hormone substitution therapy remains the mainstay treatment. | |
|
Keywords:
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, Primary amenorrhea, Turner syndrome
| |
|
Introduction
| ||||||
|
Turners syndrome is one of the most common chromosomal anomalies in humans with reported incidence of 1 in 2500 live female births [1]. It represents an important cause of primary amenorrhea with short stature, with variable hypogonadism depending the degree of gonadal mal-development. It results from complete or partial absence of the second sex chromosome with or without cell line mosaicism [2]. On the other hand, Mayer-Rokitansky-Küster-Hauser syndrome is a specific type of mullerian duct malformation characterized by the congenital aplasia of the uterus and upper two-thirds of the vagina in women with normal 46 XX karyotype [2][3]. Patients are phenotypically normal females with functioning ovaries and usually present with primary amenorrhea. Its incidence is 1 in 4500 birth [4]. The coexistence of both conditions is exceptional and probably coincidental although some theories have been reported in literature. | ||||||
|
Case Report
| ||||||
|
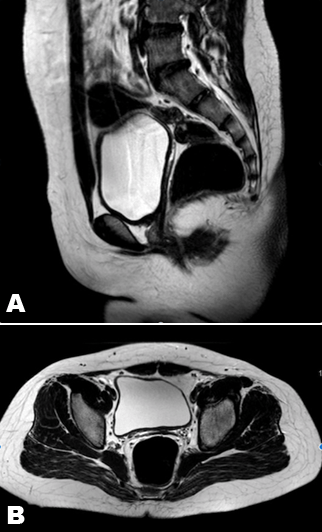
A case of 19-year-old Malay female presented with primary amenorrhea and short stature. Her parents have no consanguineous relationship with no other affected family members. Her body mass index was 22 kg/m2 with height 139 cm and weight 43 kg. On examination, her blood pressure was 130/80 mmHg. There was no discrete facial dysmorphism, neck webbing or skeletal anomalies. Based on the Tanner staging of puberty, her breast development was in Tanner stage II while pubic hair was in stage 1 (B2P1). A vaginal opening was seen, but only able to admit a cotton butt-stick to the depth of 2 cm. Plasma follicle-stimulating hormone (FSH) and luteinzing hormone levels were in postmenopausal range at 59.0 IU/L and 17.4 IU/L respectively, while serum estradiol was low, with plasma level 42.7 pmol/L (normal range for follicular phase: 0–587.0 pmol/L, for luteal phase 101.0–905.0 pmol/L). Her uterus and ovaries could not be visualized via transabdominal ultrasound. Magnetic resonance imaging (MRI) scan revealed an absent vagina, cervix and uterus, (Figure 1) consistent with diagnosis of Mullerian duct agenesis or Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. A dual energy X-ray absorptiometry (DEXA) scan showed osteoporosis changes on lumbar spine L1 to L4 and head of femur. Standard 30 cell karyotyping revealed 45, X(11)/46, X, i(X)(qter q10 :: q10 qter) consistent with Turner syndrome. Diagnosis was explained to patient and her parents. Psychological assessment by psychologist showed no emotional disturbance in the patient and parents. Conjugated equine estrogen and alendronate sodium were started to replace her estrogen and osteoporosis treatment respectively. She is currently under our adolescent gynecology clinic follow-up. | ||||||
|
Discussion
| ||||||
|
In some girls presenting with pubertal delay, features of Turner syndrome might not be so obvious, particularly with chromosomal mosaicism, hence a karyotype should be performed. Up to 25% of girls with Turner syndrome enter puberty spontaneously, however only 10% progress through puberty and only 1% develops ovulatory cycles. It is the chromosomal abnormality that seems to cause premature ovarian exhaustion rather than a primary failure of ovarian development. In its most severe form, the XO genotype patients have characteristic features including short stature, cubitus valgus, genu valgum, wide spaced nipples, cardiac, autoimmune and renal disorders. As in our case, we will not able to diagnose turner syndrome without karyotyping [5] [6] Patients with Mayer-Rokitansky-Kuster-Hauser syndrome on the other hand have a normal 46,XX genotype and normal female phenotype with spontaneous development of secondary sexual characteristic due to the presence of normal functioning ovaries. The main problem is the failure of Mullerian duct fusion, resulting in vaginal agenesis [6]. Most of them present with primary amenorrhea, and may have associate renal tract and skeletal abnormalities. With the presence of normal functioning ovaries, they do not need hormonal treatment. When appropriate, non-surgical treatment such as vaginal cone training and reconstructive surgery would enable patient to lead a normal sexual life but sadly, pregnancy is unlikely. Theoretically, ovarian stimulation followed by oocyte retrieval can be performed to achieve pregnancy via a gestational surrogate but very difficult and rare due to lack of technology or religion issue. Coexistence of both gonadal dysgenesis and MRKH has been reported, but rare. There are 26 cases were reported in literatures, 15 had normal 46, XX karyotype while the remaining had mosaicism or microdeletions [7]. Although rare, coexistence of both MRKH syndrome and gonadal dysgenesis could be explained via three theories suggested [4][7]. The first suggests that genes related to germ cells and Mullerian derivatives undergo mutation or deletion thus affecting its development and migration. The second suggests that gonads and Mullerian structure development are interrupted due microdeletion on the X chromosomes leading to absence of certain proteins required for its formation. The third hypothesis suggests that the role of endocrine mediators but very few cases have been published with regards to it. Early diagnosis of MRKH syndrome and Turner syndrome is very difficult. As in our patient, early diagnosis and treatment might prevent complications such as short stature and osteoporosis. Psychological impacts such as low self-esteem and concern of femininity might affect her in future. Therefore, psychological counseling is one of the vital component in managing this patient. Gender identity is not an issue because she is phenotypically female. Estrogen replacement therapy is needed to promote her secondary sexual characteristics especially the breast development. Alendronate sodium can prevent further osteoporosis. Sexual function is not an issue at this moment because she is not sexually active. She might benefit from vaginal cone traning or vaginal reconstructive surgery in the future. Sadly, pregnancy is very unlikely possible for her due to coexistence of Turner syndrome and MRKH syndrome. | ||||||
| ||||||
|
| ||||||
|
Conclusion
| ||||||
|
The association of Turner syndrome and Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is extremely rare and appears to be coincidental, independent of chromosomal anomalies. The target of the treatment is to promote the development of secondary sexual characteristics, to prevent osteoporosis and psychological support. Multidisciplinary team approach is the main key of management of this patient. | ||||||
|
References
| ||||||
| ||||||

|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Nik Rafiza Afendi – Substantial contributions to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Hoo Ps – Substantial contributions to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Mas Irfan Jaya Mahamooth – Substantial contributions to conception and design, Analysis and interpretation of data, Drafting the article, Final approval of the version to be published Ahmad Amir Ismail – Substantial contributions to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Rahimah Abdul Rahim – Substantial contributions to conception and design, Acquisition of data, Drafting the article, Final approval of the version to be published Ahmad Akram Omar – Substantial contributions to conception and design, Acquisition of data, Drafting the article, Final approval of the version to be published Adibah Ibrahim – Substantial contributions to conception and design, Acquisition of data, Drafting the article, Final approval of the version to be published |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2017 Nik Rafiza Afendi et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|